 15 września 2022
15 września 2022  Renata Morawska-Wyłupek
Renata Morawska-Wyłupek
 Arvato
Arvato

Serializacja wyrobów medycznych, a aspekty prawne
Serializację wyrobów medycznych regulują następujące dokumenty:
- Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2017/745 z dnia 5 kwietnia 2017 r. w sprawie wyrobów medycznych.
- Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2017/746 z dnia 5 kwietnia 2017 r. w sprawie wyrobów medycznych do diagnostyki in vitro oraz Ustawa z dnia 7 kwietnia 2022 r. o wyrobach medycznych, która precyzuje wdrożenie powyższych na rynku polskim.
Oba rozporządzenia Parlamentu Europejskiego dotyczą regulacji prawnych i obowiązku zabezpieczeń wyrobów medycznych dla całej Unii Europejskiej. Rozporządzenia opisują zakres zabezpieczeń wyrobów medycznych, począwszy od ich produkcji, skończywszy na dotarciu do odbiorcy. W obu rozporządzeniach zawarty jest artykuł 14, określający obowiązki dystrybutorów.
Najważniejszymi zadaniami firm logistycznych są kontrole zabezpieczeń i oznakowania opakowań oraz zapewnienie wymaganych warunków dla przechowywania i transportu wyrobów. Ustawa z dnia 7 kwietnia 2022 r. o wyrobach medycznych przedstawia normy dotyczące udostępniania wyrobów medycznych i wprowadzania ich do użytku powszechnego na terenie naszego kraju. Prawo reguluje również wytyczne dotyczące logistyki wyrobów medycznych, które możemy znaleźć w rozdziale 13 – Używanie i utrzymanie wyrobów medycznych. Przepis nakłada obowiązek posiadania odpowiedniej dokumentacji oraz spełnienia wszystkich norm dotyczących dystrybucji i magazynowania wyrobów. Ustawa określa obowiązki podmiotów gospodarczych, instytucji zdrowia publicznego, podmiotów wykonujących działalność leczniczą, osób wykonujących zawody medyczne oraz innych podmiotów, właściwość, uprawnienia, obowiązki i zadania organów, a także kary administracyjne, zasady i tryb prowadzenia badania klinicznego wyrobu medycznego i badania działania wyrobu medycznego do diagnostyki in vitro, wysokość i tryb uiszczania opłat.
Większość producentów wyrobów medycznych, w całym łańcuchu dystrybucyjnym, wymaga od swoich dostawców i podwykonawców wykazania zgodności z normą ISO 13485 w zakresie systemów jakości dedykowanych wyrobom medycznym, spójnych z wymaganiami ściśle powiązanymi z branżą farmaceutyczną.
Czym jest wyrób medyczny?
Już w samych rozporządzeniach możemy znaleźć informację, że definicja wyrobu medycznego jest dość szerokim pojęciem i często wymaga odrębnych interpretacji. Wyroby medyczne to zatem:
- narzędzia,
- przyrządy,
- urządzenia,
- oprogramowanie komputerowe,
oraz materiały lub inne artykuły, które mogą być stosowane samodzielnie lub w połączeniu do użytku u ludzi do co najmniej jednego z następujących szczególnych zastosowań medycznych. Wśród najważniejszych celów znajduje się: diagnozowanie, zapobieganie, monitorowanie, leczenie lub łagodzenie przebiegów chorób, urazów czy upośledzeń.
Prócz tego wyrobem medycznym może być również badanie, zastępowanie lub modyfikacja budowy anatomicznej ciała, proces fizjologiczny czy narzędzia do regulacji poczęć. Tak obszerna definicja wymaga szeregu regulacji prawnych, które na każdym etapie dają możliwość kontroli i zagwarantowania najwyższego bezpieczeństwa.[1]
Czym jest serializacja?
Serializacja, w znaczeniu farmaceutycznym, odnosi się w głównej mierze do leków na receptę. W przypadku leków bez recepty, suplementów czy większości wyrobów medycznych, taki proces nie jest wymagany. Warto jednak wspomnieć, że serializacja to podstawowy i najważniejszy etap bezpośredniego zabezpieczania produktów leczniczych, który obecny jest na każdym etapie łańcucha dostaw.
W wyrobach medycznych, w rozumieniu art. 2 pkt 15 rozporządzenia 2017/745 lub art. 2 pkt 15 rozporządzenia 2017/746, serializacja odnosi się do zastosowania tzw. kodów UDI (Niepowtarzalny Kod Identyfikacyjny Wyrobu). Mają one na celu prostą, jednoznaczną identyfikację konkretnego wyrobu medycznego na etapie jego dystrybucji i użytkowania. Sekwencja znaków numerycznych i alfabetycznych, czytelnych dla człowieka (w postaci zwykłego tekstu) jak i odczytu maszynowego (AIDC) dzieli kod na UDI-PI i UDI-DI. Kod UDI-PI ma na celu wskazanie jednostki produkcyjnej wyrobu. Zawiera on numer serii lub numer seryjny, identyfikację oprogramowania oraz datę ważności. Kod UDI-DI to kod Basic UDI-DI wyrobu zawierający informację na temat producenta jak i samego indywidualnego wyrobu. [2]
Serializacja – kod UDI
Obowiązek umieszczania kodu UDI na wszystkich poziomach opakowań oraz na etykietach stosuje się lub będzie się stosować m.in. w przypadkach:
- wyrobów do implantacji i wyrobów klasy III (wysokiego ryzyka) takich jak np. zastępcze zastawki serca, protezy kolan, baterie do rozruszników serca (konieczność stosowania od dnia 26 maja 2021 roku),
- wyrobów klasy IIa i klasy IIb (umiarkowane ryzyko) takich jak np. opatrunki hydrożelowe, cewniki jednorazowe, klisze rentgenowskie, pojemniki na krew, prezerwatywy, respiratory (konieczność stosowania od 26 maja 2023 roku),
- wyrobów klasy I (niskie ryzyko) takich jak np. wyroby sterylne, rękawice do badań, kołnierze ortopedyczne, wózki inwalidzkie, wyroby z funkcją pomiarową (konieczność stosowania od 26 maja 2025 roku),
- wyrobów medycznych wielokrotnego użytku, które muszą zawierać nośniki kodu UDI na samym wyrobie (konieczność stosowania po upływie 2 lat od daty wynikającej z klasy wyrobu),
- wyrobów medycznych do diagnostyki in vitro klasy D (konieczność stosowania od 26 maja 2023 roku),
- wyrobów medycznych do diagnostyki in vitro klasy B i C (konieczność stosowania od 26 maja 2025 roku),
- wyrobów medycznych do diagnostyki in vitro klasy A (konieczność stosowania od 26 maja 2027 roku) [3].

EUDAMED
Pisząc o identyfikacji wyrobów medycznych należy również wspomnieć o EUDAMED, czyli Europejskiej Bazie Danych o Wyrobach Medycznych. Będzie to jedno z kluczowych narzędzi wprowadzonych na mocy rozporządzenia Parlamentu Europejskiego i rady UE. W skład bazy wejdzie sześć modułów, których zadaniem będzie:
- Elektroniczny system rejestracji podmiotów gospodarczych,
- Elektroniczny system rejestracji wyrobów medycznych i kodów UDI,
- Elektroniczny system dotyczący jednostek notyfikowanych i wydawanych przez nie certyfikatów,
- Elektroniczny system dotyczący nadzoru rynku,
- Elektroniczny system dotyczący obserwacji i nadzoru po wprowadzeniu do obrotu wyrobu medycznego,
- Elektroniczny system dotyczący badań klinicznych.[4]
Dzięki stosowaniu kodów UDI nie tylko mamy możliwość identyfikacji wyrobów, ale również i ich łatwiejszego wycofania i zwalczania nielegalnych podróbek, a co najważniejsze, poprawy bezpieczeństwa pacjentów.
Projektowanie przestrzeni magazynowej dla wyrobów medycznych. Must-have i nice-to-have
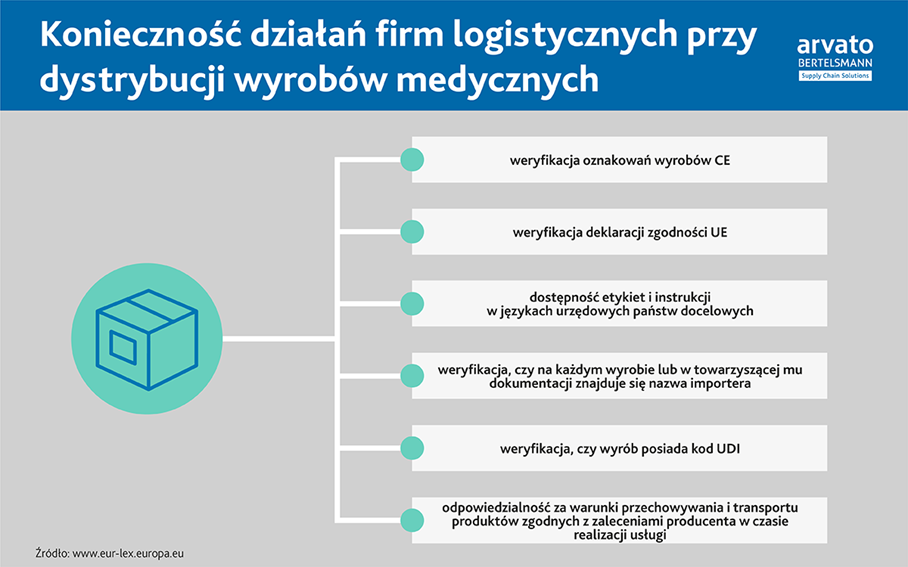
Firmy logistyczne jako dystrybutorzy w łańcuchu dostaw są zobligowani do następujących działań:
- weryfikacji oznakowań wyrobów CE,
- weryfikacji deklaracji zgodności UE,
- dostępności etykiet i instrukcji w językach urzędowych państw docelowych,
- weryfikacji, czy na każdym wyrobie lub w towarzyszącej mu dokumentacji znajduje się nazwa importera,
- weryfikacji, czy wyrób posiada kod UDI,
- odpowiedzialności za warunki przechowywania i transportu produktów zgodnych z zaleceniami producenta w czasie realizacji usługi.[5]
Elementami, które nie są konieczne pod względem prawa, lecz bez wątpienia niezbędne przy logistyce wyrobów medycznych, są systemy śledzenia zamówień oraz kontroli stanów magazynowych. To dzięki nim zapewnimy wyrobom medycznym dodatkowe bezpieczeństwo. Dzięki automatycznemu systemowi znakowania oraz zarządzania projektowaniem i wydrukiem etykiet, mamy możliwość zoptymalizowania i przyspieszenia procesów magazynowych.
Projektując przestrzeń magazynową warto pamiętać również o odpowiednim podziale na strefy i zapewnieniu sprawnej komplementacji zamówień o niewielkich rozmiarach.
Wyzwania operatorów logistycznych w branży medycznej
W ostatnich latach operatorzy logistyczni, oprócz codziennych wyzwań projektowych, jakimi są restrykcyjne kontrole jakości, specjalne warunki magazynowania i transportu czy ścisłe terminy dostaw, stanęły przed wyzwaniem zaspokojenia ogromnego zapotrzebowania na wyroby medyczne.
Pandemia oraz działania wojenne w Ukrainie zmieniły rynek farmaceutyczny. Firmy farmaceutyczne zwiększyły wydajność produkcji i tym samym wprowadziły zmiany w logistyce wyrobów medycznych. W przestrzeniach magazynowych koniecznością jest nierzadko przyjęcie większej liczby produktów oraz szybkie dostosowanie warunków do przechowywania nowych elementów, ale także ulepszenie procesów dystrybucyjnych. Zamknięcie największych portów dystrybucyjnych oraz przestrzeni powietrznej spowodowało ryzyko przerwania łańcucha dostaw. To wymogło konieczność strategicznego planowania i tym samym priorytetyzację produkcji, oszacowanie zapotrzebowania na wyroby oraz konieczność zwiększenia liczby pracowników poprzez wprowadzenie systemu trzyzmianowego.
Wielu z nas nie jest świadomych, że dzięki szybkiej reakcji rynku logistycznego mieliśmy stały dostęp do najpotrzebniejszych leków jak i wyrobów medycznych, a ciągłe doskonalenie procesów logistycznych oraz wprowadzanie nowych technologii (w tym automatyzacji) do przestrzeni magazynowych. Pomogło to zoptymalizować procesy magazynowe, a tym samym skrócić czas dostaw produktów ratujących życie.
[1] Źródło: https://www.urpl.gov.pl/pl/wyroby-medyczne/wprowadzenie-wyrob%C3%B3w-medycznych-do-obrotu-i-do-u%C5%BCywania/informacje-dotycz%C4%85ce-0
[2] Źródło: https://eur-lex.europa.eu/legal-content/PL/TXT/PDF/?uri=CELEX:32017R0745&from=IT
[3] https://www.wyrobmedyczny.info/wyroby_medyczne/kody-udi/
[4] https://www.wyrobmedyczny.info/wyroby_medyczne/eudamed/
[5] Źródło: https://eur-lex.europa.eu/legal-content/PL/TXT/?uri=celex%3A32017R0745